第二十七章 免疫缺陷病的检验
由于遗传或其他原因造成的免疫系统先天发育不全或获得性损伤称为免疫缺陷(immunodeficiency);由此所致的各种临床征称为免疫缺陷病(immunodeficiencydiseases)。免疫缺陷病的主要临床特征是病人有反复延性或机会性感染,易发生恶性肿瘤,常伴发过敏性疾病和自身免疫病。
第一节 免疫缺陷病的发病机制
一、免疫系统的个体发育
人体的免疫系统是在遗传控制下发育成熟的。胸腺发生于咽囊,胚胎时期干细胞发生于卵囊,以后逐渐分化为各系的祖细胞并分别沿各自的途径分化成熟。例如在胸腺内分化成熟的为T细胞,B细胞则在骨髓中直接成熟。
人类的免疫球蛋白最早出现在胚胎第10~20周,最先出现的是IgM,IgG和IgA次之。至出生时IgM可达成人水平的10%;IgA,1.2%;IgG,82%;后者包括从母体传输过来的一大部分。测定儿童免疫球蛋白水平是评价其免疫功能的一种重要方法。0~15岁儿童血清免疫球蛋白平均水平见表27-1。表中数字为占成人Ig水平的百分比。
表27-1 各年龄组儿童的血清Ig相对水平
| 年龄组 | IgG | IgM | IgA |
| 新生儿(脐带血) | 82 | 10 | 1.2 |
| 2~7天 | 81 | 11 | 0.9 |
| 8~13天 | 79 | 46 | 2.4 |
| 31~60天 | 52 | 35 | 5.2 |
| 3~4月 | 38 | 54 | 6.9 |
| 5~6月 | 38 | 60 | 8.8 |
| 7~12月 | 56 | 82 | 16 |
| 1~2岁(13~24月) | 61 | 86 | 25 |
| 3~4岁(25~36月) | 60 | 91 | 29 |
| 4~5岁 | 69 | 82 | 40 |
| 6~8岁 | 87 | 106 | 79 |
| 9~11岁 | 90 | 96 | 78 |
| 12~15岁 | 92 | 113 | 95 |
| 成年人 | 100 | 100 | 100 |
细胞反应最早出现在胚胎第12~15周,但至出生时也远不成熟,迟发型反应较弱。新生儿的吞噬功能也不完备,吞噬细胞在数量和功能上均未过成人水平。补体发育比较早一些,至出生时补体的血清水平可达成人的50%,旁路途径也已成熟。
二、免疫缺陷病的发病机制
(一)免疫缺陷的发生阶段及环节
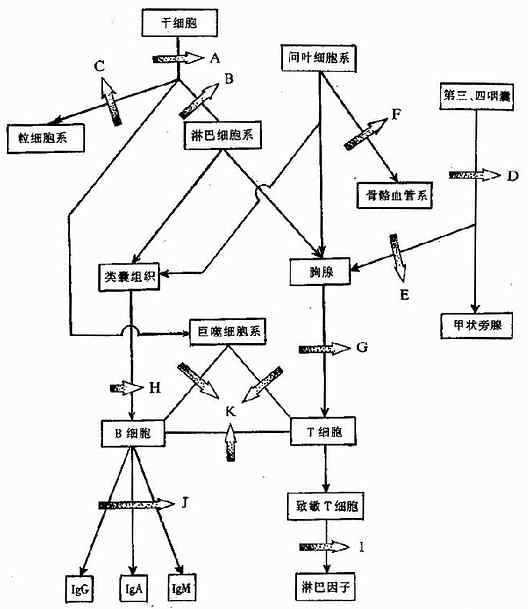
免疫系统在发生和发育过程中,由于某种原因导致某个环节受阻,就会发生免疫缺陷(图27-1)。
1.干细胞发育障碍(图27-1A)使淋巴细胞系、粒细胞系及巨核细胞系皆可发生缺陷。
2.淋巴细胞系发育障碍(图27-1B)形成联合免疫缺陷病。
3.粒细胞发育障碍(图27-1C)可发生慢性肉芽肿。
4.第三、四咽囊形成障碍(图27-1D)使胸腺和甲状旁腺发育不良,常见的疾病有DiGeorge综合征等。
5.胸腺形成不良(图27-1E)可导致Nezelof综合征。
6.间叶细胞系发育障碍(图27-1F)骨髓血管系异常,可出现共济失调性毛细血管扩张症等。
7.胸腺功能障碍(图27-1G)T细胞异常,可导致共济失调性毛细血管扩张症等。
8.类囊组织的障碍(图27-1H)影响B细胞形成,可出现性连无丙种球蛋白血症。
以上皆为中枢性干细胞发育障碍所致的免疫缺陷。
图27-1 免疫系统各阶段发育障碍示意图
(A~K为发育障碍的部位)
9.致敏T细胞功能不全(图27-1I)可发生慢性皮肤粘膜真菌感染症等。
10.B细胞至浆细胞阶段发生障碍(图27-1J)可发生选择性免疫缺陷病,例如IgA选择性缺陷病等。
11.T细胞和B细胞协同作用障碍(图27-1K)也是一种联合免疫缺陷,但属于外周性的,例如Wiskott-Aldrich综合征等。
以上各种免疫缺陷的发生率相差很大,其中B细胞缺陷最多见(约50%),其次为联合免疫缺陷(20%)(图27-2)。
(二)免疫缺陷的发生原因
1.遗传基因异常一是X连锁隐性遗传,病态基因位于X染色体上,在女性可以不表现疾病,但在男性则别无选择;所以男性患病率比女性高得多;例如Bruton型丙种球蛋白缺乏症、X连锁高IgM型Ig缺陷症和X连锁联合免疫缺陷病(Gitlin型)等。另一类为常染色体隐性遗传,发病无性别差异,例如Swiss型联合免疫缺陷病等。
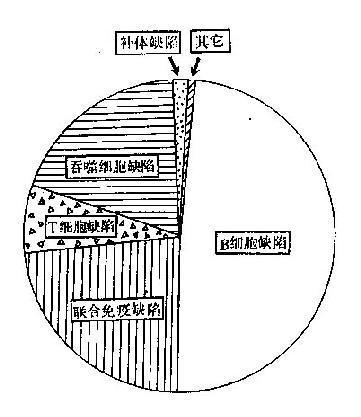
图27-2 各型免疫缺陷的发生率
2.中枢免疫器官发育障碍可由遗传缺陷所致,也可由宫内感染某些病毒(例如巨细胞病毒、麻疹病毒等)导致胚胎发育受损,免疫系统发育异常。
3.免疫细胞内在缺陷多由先天性酶缺陷引起,例如腺苷脱氨酶(ADA)缺乏、核苷磷酸化酶(PNP)缺乏以及葡萄糖-6-磷酸脱氢酶(G6P-D)缺乏皆可引起T细胞、B细胞或吞噬细胞缺陷。
4.免疫细胞间调控机制异常机体的免疫调控是一个极为复杂的网络机制,在这个调节网中,无论是辅助不足或抑制过盛,皆可导致免疫缺陷。许多继发性免疫缺陷则常因感染、药物和放射线等因素而发生。